Interactive Analaysis GUI
Author:Andrew Causer
This notebook is a tutorial of how to use the interactive analysis GUI implmeneted in goatpy. This interactive GUI is build using Napari, for more infomation on how to use napari please visit here.
For more infomation on how to run general analysis using goatpy please visit the Getting Started page.
Introduction to goatpy GUI
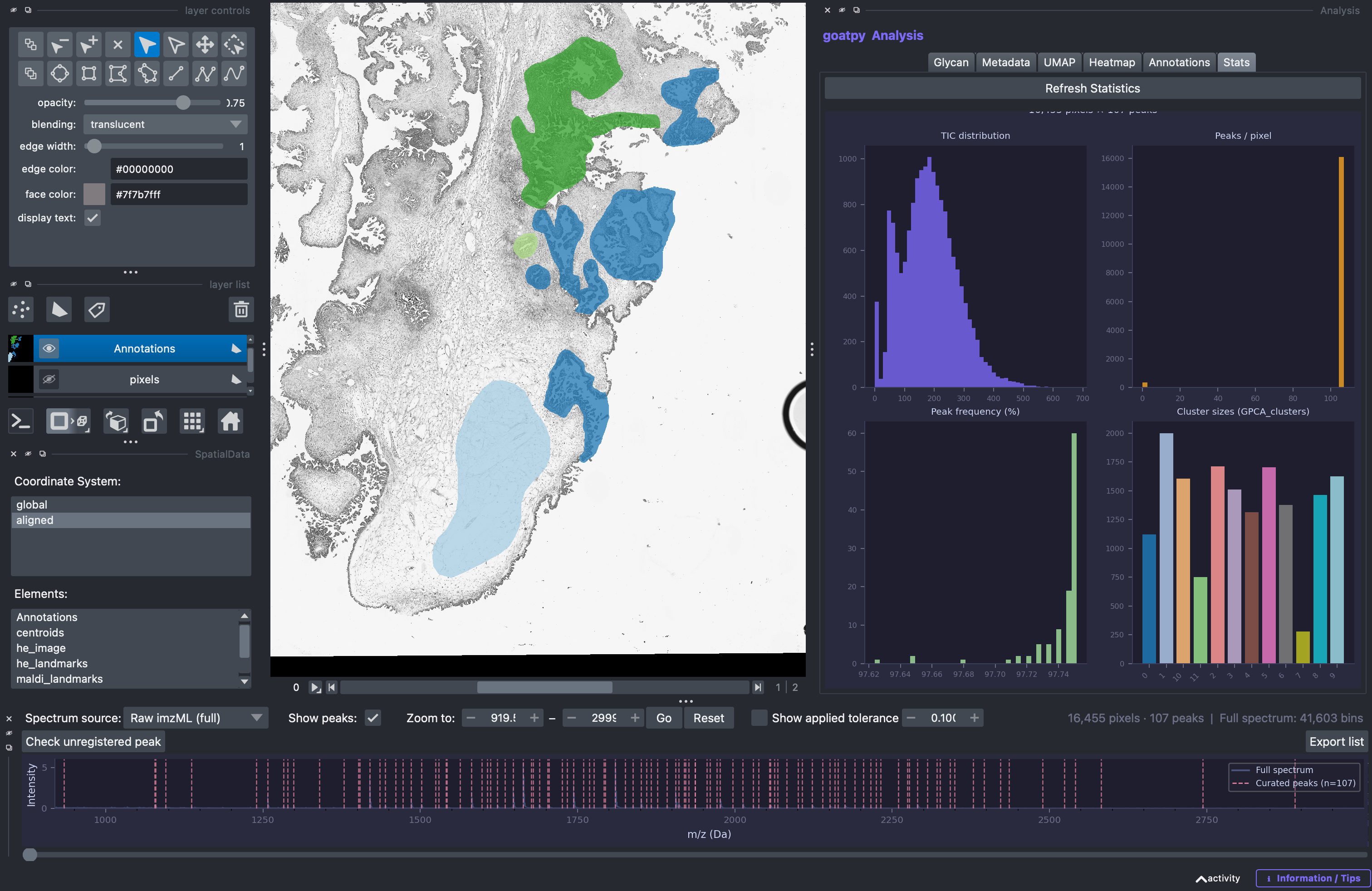
The goatpy GUI is made up of 4 main panels:
Center: View window containing the spatial glycomics dataset
Left (‘Layer Control’, ‘Layer List’, ‘SpatialData’):
NapariandSpatialDatatools for selecting elements to visualise, corresponding coordinate systems and for additional napari tools such as polygon drawing (i.e. ROI, Pathologist Annotations, etc.).Right (‘Analysis’):
goatpyAnalysis window containing a range of tools including, glycan visualisation, clustering UMAPs, heatmap generation, Annotation editing and metadata plotting.Bottom (‘Spectrum’): Spectra panel contating the MSI spectra highlighting loaded peaks, successfully named glycans and more
Each panel has various applications which can assist across the analysis pipeline.

Once loaded you will see a window looking something like this, which contains the dataset along with the 4 main panels described above.
Changing GUI Layout
Panels can be hidden or viewed by heading to the menu bar and selecting window where a list of each panel will be available to select or unselect to improve visualisation on smaller screens.
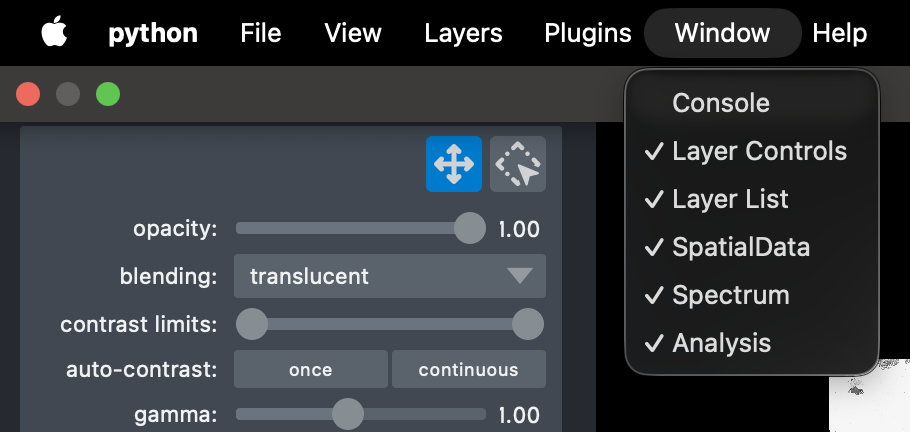
Alternatively, each widget can be maximised into it’s own window and moved, for an adjustable layout. Note: double clicking the outer window bar will return it to inside the main windown.
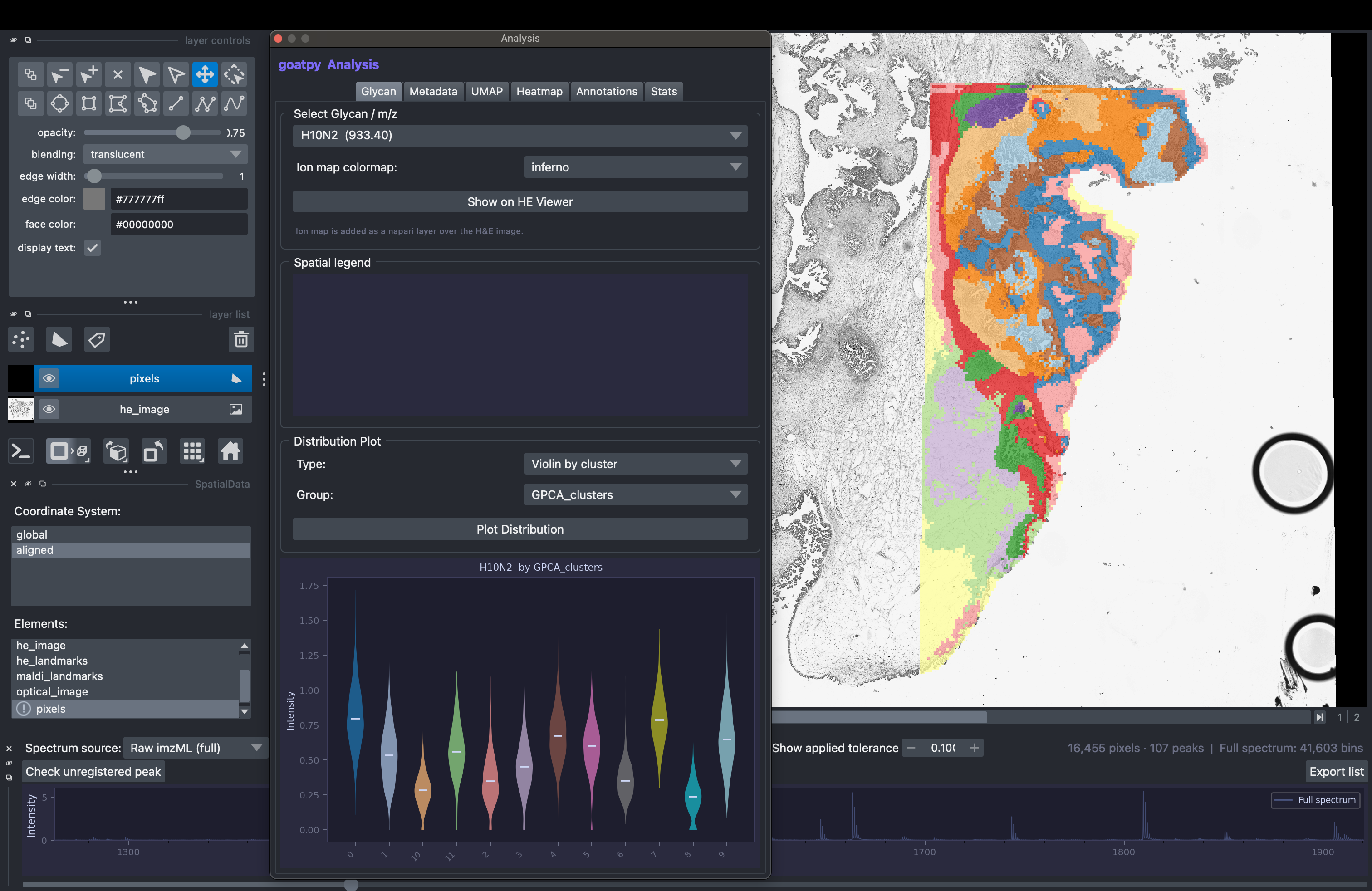
For more infomation on how each panel works there is an infomation/tips tab located in the bottom right corner.
Pre-Processing: Peak Selection
Using goatpy we can read in our .imzml files and create SpatialData objects. These objects can then be visalised with goatpy's napari-based viewer to assess the loaded data before downstream analysis. This is particularly good for:
Adding additional m/z peaks to the dataset that were not included in the original peak list used for loading
Adjusting loaded tolereance value for peak binning
Annotating ROI’s for large datasets
Generate quality checking and data exploration
First we will load the goatpy library and relative raw data.
[1]:
import goatpy as gp
[2]:
raw_data = gp.glyco_spatialdata(imzml_path="./Documents/AD_resolution_change_check_10um_actual_10012020_2.imzML")
Once our data is loaded we can run the goatpy GUI to visualise the data.
[3]:
viewer = gp.launch_goatpy_gui(raw_data)
2026-06-28 23:00:56.523 | WARNING | napari_spatialdata._viewer:__init__:57 - Due to Shift-L being used as shortcut in napari, it is being deprecated and might not link a new layer to an existing SpatialData object in the viewer. Please use ⌘-L on MacOS or else Ctrl-L.
[goatpy GUI] Added 'pixels' (shapes) in 'global'
INFO: Please wait for the current operation to finish.
[goatpy GUI] Added 'centroids' (points) in 'global'
[goatpy GUI] Could not switch coordinate system: aligned not present as coordinate system in any of the spatialdata objects
[goatpy GUI] Ready
16455 pixels · 108 peaks
WARNING: QStatusBar::insertPermanentWidget: Index out of range (0), appending widget
2026-06-28 23:01:02.345 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-06-28 23:01:02.358 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
INFO: Layer updated: 933.4000 (933.40)
INFO: Layer updated: 1809.6560 (1809.66)
INFO: Layer updated: m/z 1978.7943 (±0.5000)
INFO: Ion image displayed for m/z 1978.7943 (m/z 1978.7943 ± 0.5000)
INFO: Added m/z 1978.7943 to list (n=109).
Once loaded you will see a window which contains the dataset you have loaded:
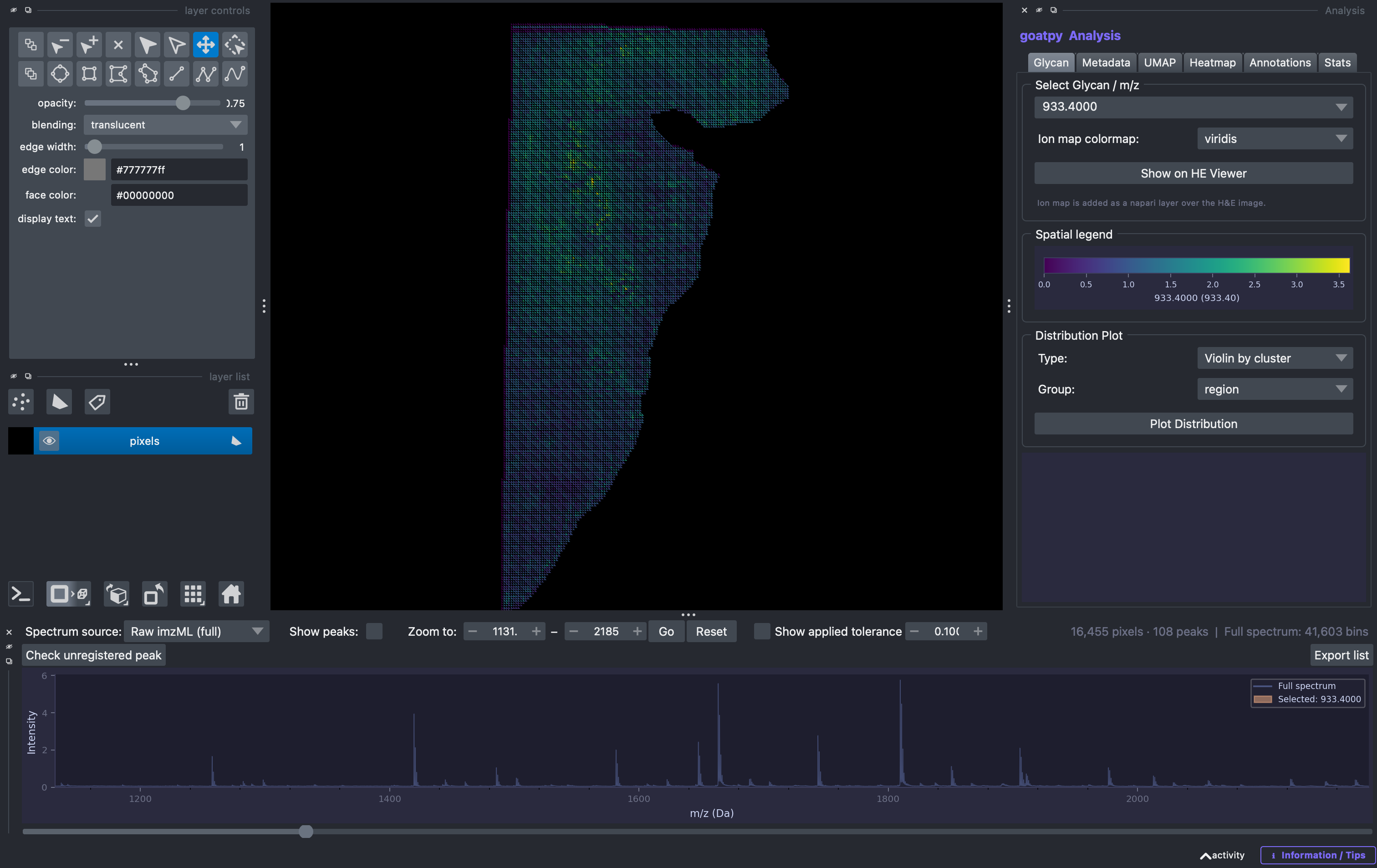
For the purpose of checking loaded m/z peaks we will focus on the ‘Spectrum’ panel at the bottom of the window. When selecting the show peaks option, all loaded peaks will be highlighted by dashed red lines indicating their relative m/z values. To visualise a peak you can either select it in the Glycan tab found in the Analysis window on the right, or simply clicking on the dashed red line in the spectral plot. The selected m/z will then be highlighted in orange:
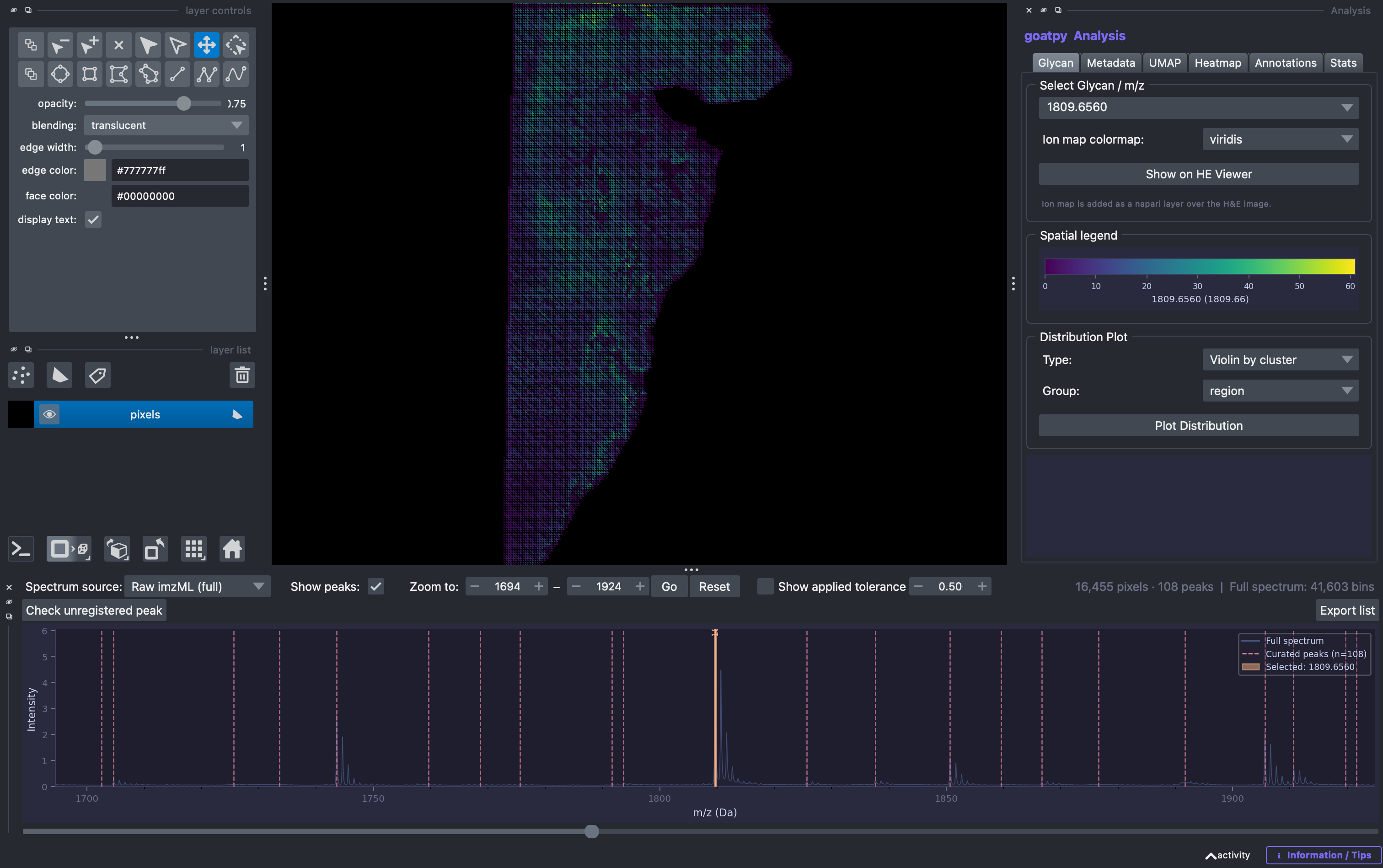
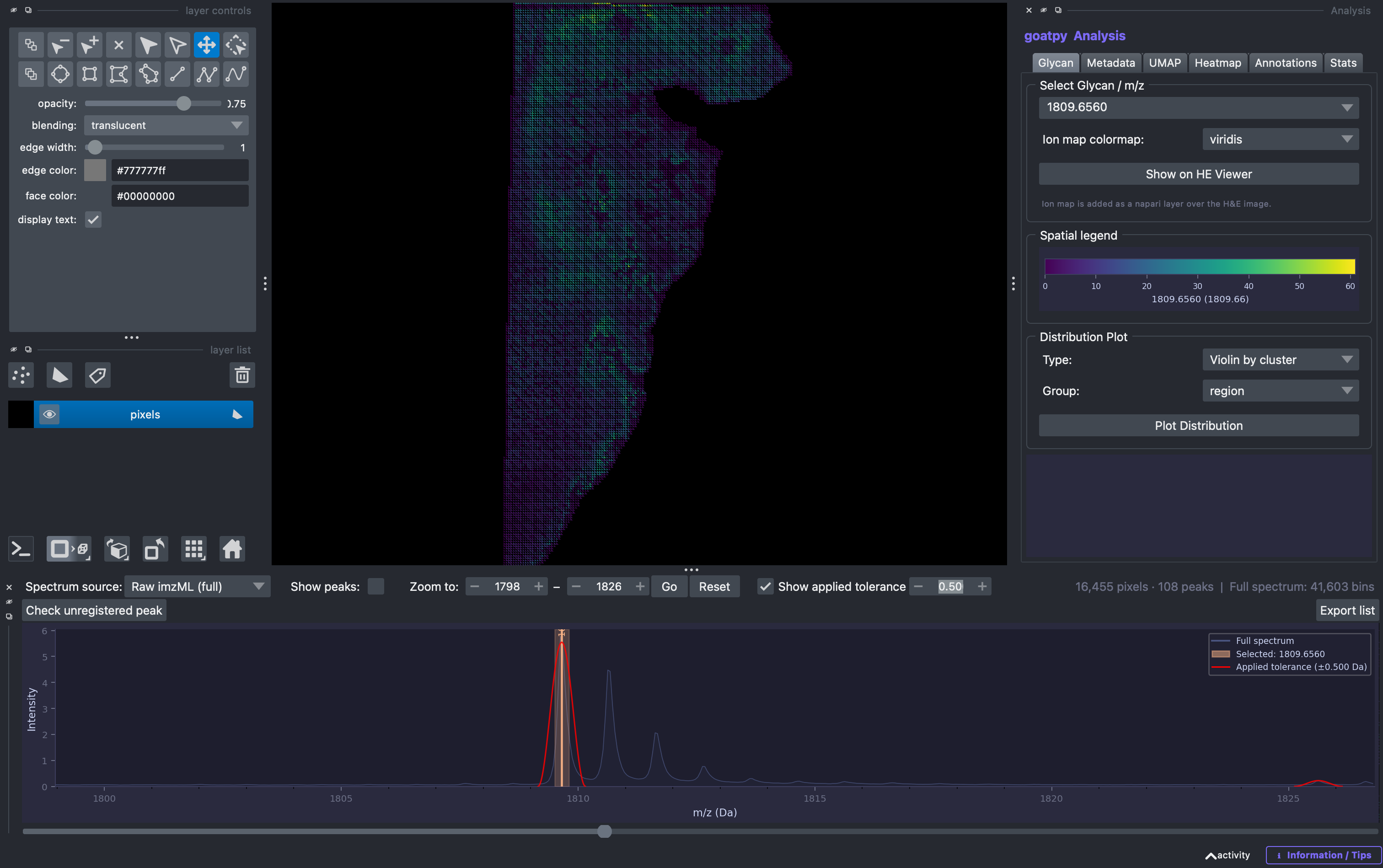
In addition the Show applied tolerence option can also be selected and adjusted to visualise the corresponding binning applied to each loaded peak, and observe how adjusting the tolerance may imporve the analysis. The relative binning curve is depicted by a red curve.
You can see that not all peaks have been loaded into this data object. This is often because they are noise and thus have not been included in the peak list used to load the data. However, some missed peaks may contain actual biological relivance and should be included in the analysis.
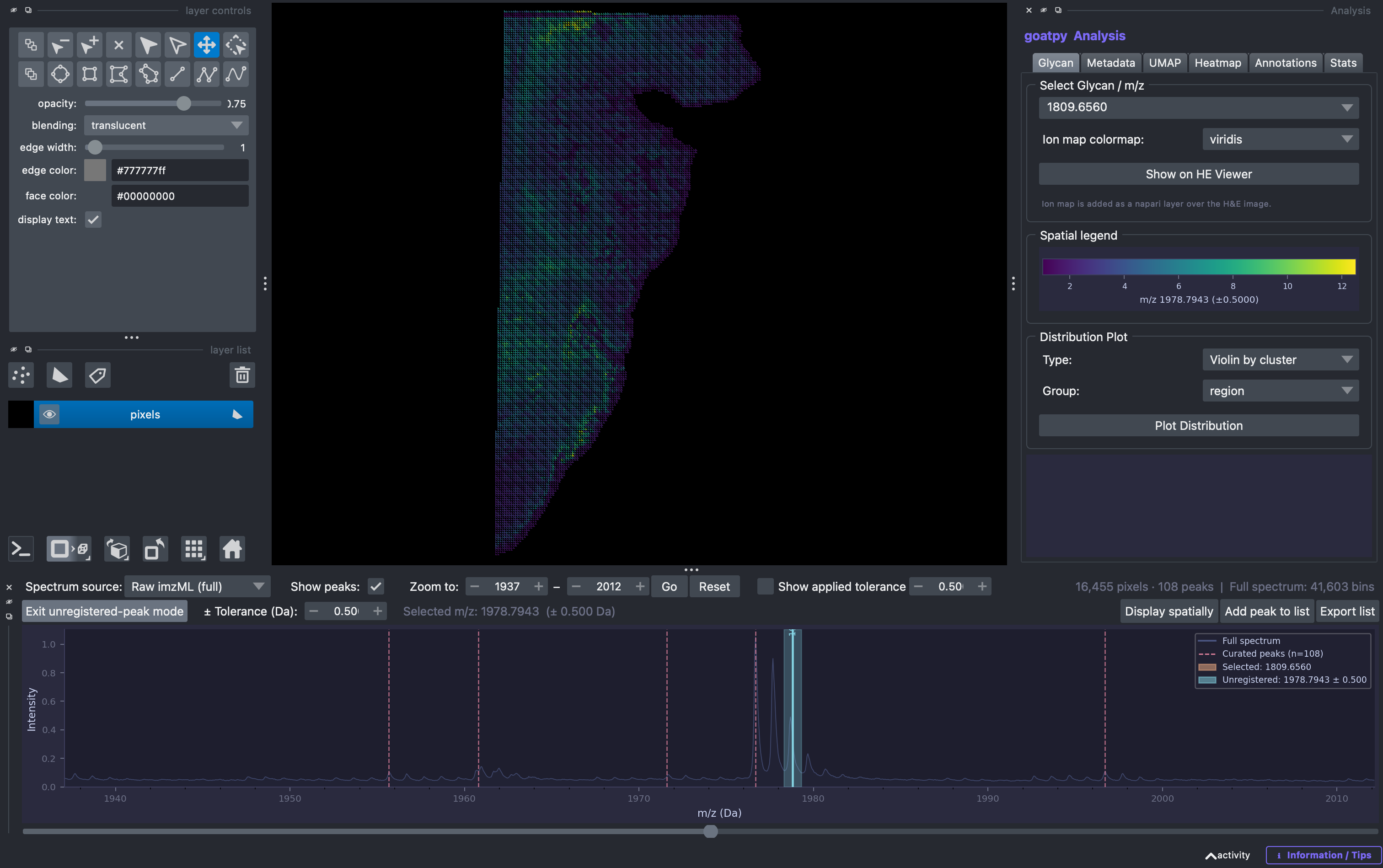
Using the Check unregisted peak button, users can select any peak that is not highlighted by a red line (inidicating that peak is already included in the dataset) on the spectrum, and visualise the spatial intensity by selecting display spatially. If you want to add this peak to the list of m/z values loaded when importing the data you can select the Add peak to list and then the export list to export a .csv file contating all original and new m/z peaks. This file can be
used to reload the raw data to contain all the relative data required for the corresponding analysis.
Post-Processing: Data Visualisation
This GUI can also be used for post-processing analysis, with a wide range of applications. We will first load a processed goatpy object (full analysis found in the Getting Started tutorial) and then use the GUI to visualise the results.
Using SpatialData pre-build data storage structure (.zarr) we can easily load our already analysed data.
[4]:
import goatpy as gp
from spatialdata import read_zarr
sdata = read_zarr("./Documents/data_objects/ovarian.zarr")
Now lets run the goatpy GUI to look at our processed data:
[5]:
viewer = gp.launch_goatpy_gui(sdata)
2026-07-01 08:12:06.303 | WARNING | napari_spatialdata._viewer:__init__:57 - Due to Shift-L being used as shortcut in napari, it is being deprecated and might not link a new layer to an existing SpatialData object in the viewer. Please use ⌘-L on MacOS or else Ctrl-L.
[goatpy GUI] Added 'he_image' (images) in 'aligned'
INFO: Please wait for the current operation to finish.
[goatpy GUI] Added 'optical_image' (images) in 'aligned'
[goatpy GUI] Added 'Annotations' (shapes) in 'aligned'
[goatpy GUI] Added 'pixels' (shapes) in 'aligned'
[goatpy GUI] Added 'centroids' (points) in 'aligned'
[goatpy GUI] Added 'he_landmarks' (points) in 'aligned'
[goatpy GUI] Added 'maldi_landmarks' (points) in 'aligned'
[goatpy GUI] Ready
16455 pixels · 108 peaks
2026-07-01 08:12:10.998 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:12:10.999 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:13:15.767 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:13:15.785 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:13:15.881 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:14:32.214 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
2026-07-01 08:14:32.218 | DEBUG | napari_spatialdata._view:_on_layer_update:569 - Updating layer.
Selecting Layers
When loading a sample with multiple layers of data it is important to select the infomation you wish to display. The SpatialData panel contains two sections - ‘Coordinates’ and ‘Elements’. For data aligned with the manual alignment apparoch the ‘Coordinates’ column will contain both ‘global’ and ‘aligned’ coordinate systems. It is important to ensure the ‘aligned’ option is selected to visualise spatial data overlayed with the H&E image (for automated alignment datasets the only
option will be ‘global’). Likewise, it is important to select the elements you want to visualise - for example:
When displaying glycan intensities the ‘pixel’ element must be selected to observe these spatially.
When displaying pathologist annotations the ‘Annotation’ element must be selected.
The selected elements should appear in the Layer List section.
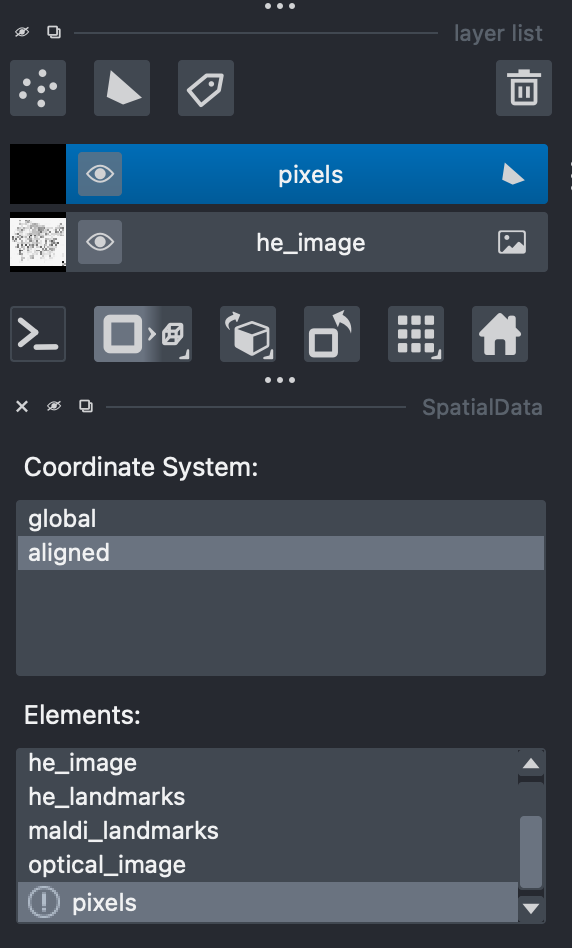
Here, the ‘pixels’ elements are loaded meaning any glycan intensities or metadata can be displayed across all pixels.
For example lets try and visualise the ‘Annotations’:
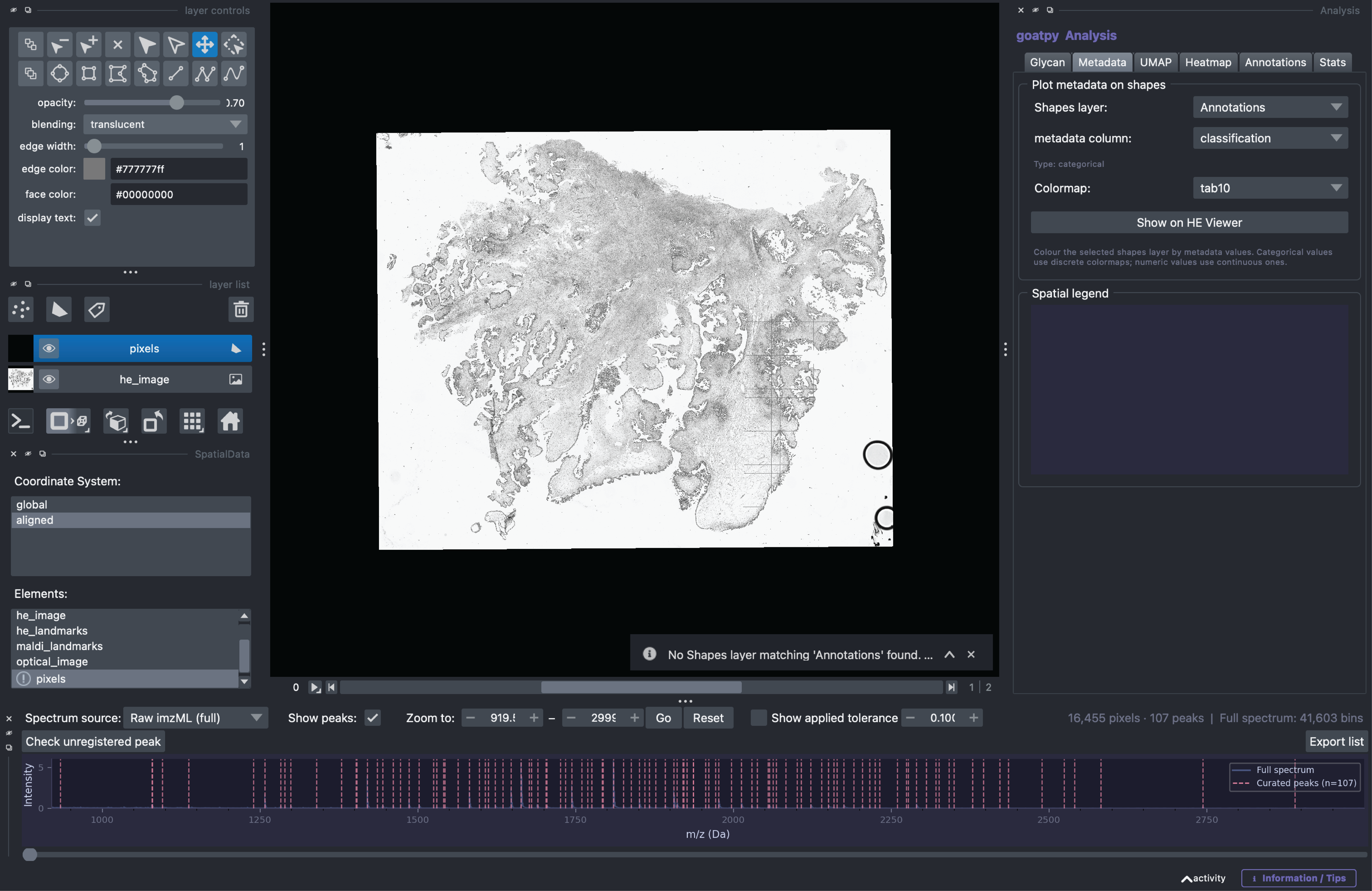
If the correct layer has not been selected you will see an error such as this:
“ℹ️ No Shapes layer matching ‘Annotations’ found …”
Then you would need to go across to the ‘Elements’ section and double click the cooresponding ‘Annotations’ element.
Glycan Plotting
After selecting the correct layers we can head over to the ‘Analysis’ window to plot a variety of different analysis results. Looking at the glycan tab first we can select any glycan present in the dataset to plot.
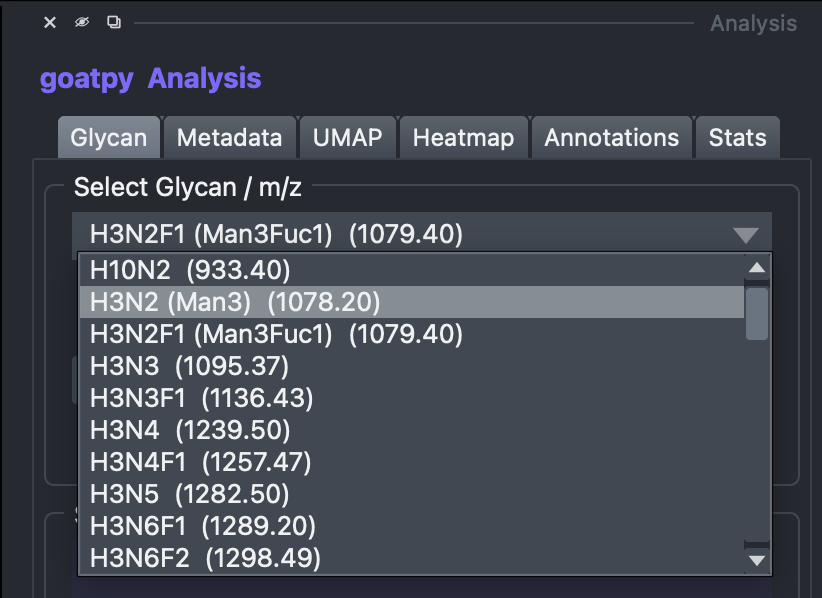
Once selected we can also change the colour map used to plot the intensity values, in this case we will select ‘jet’ and then press Show on HE Viewer to display the results spatially:
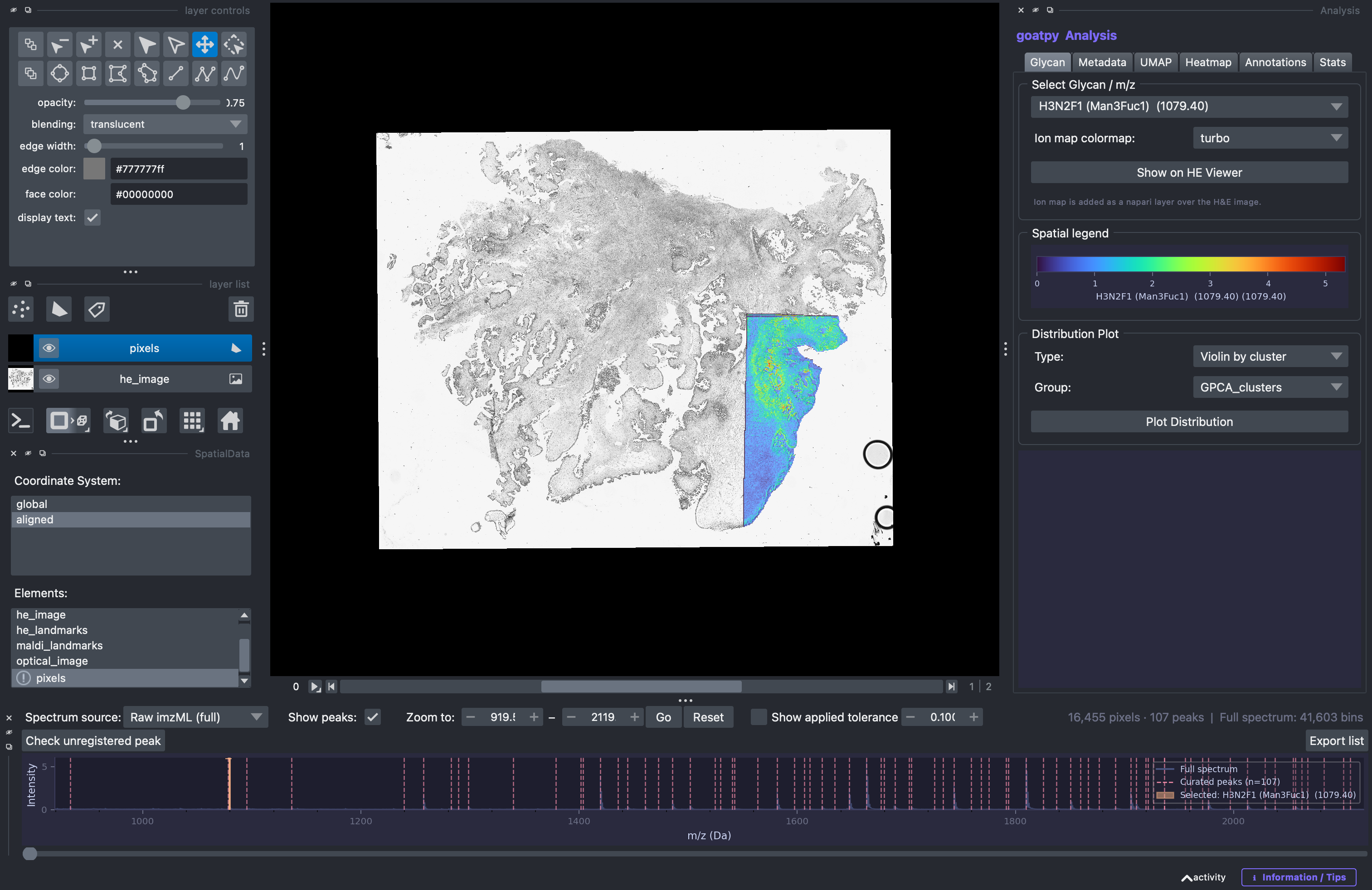
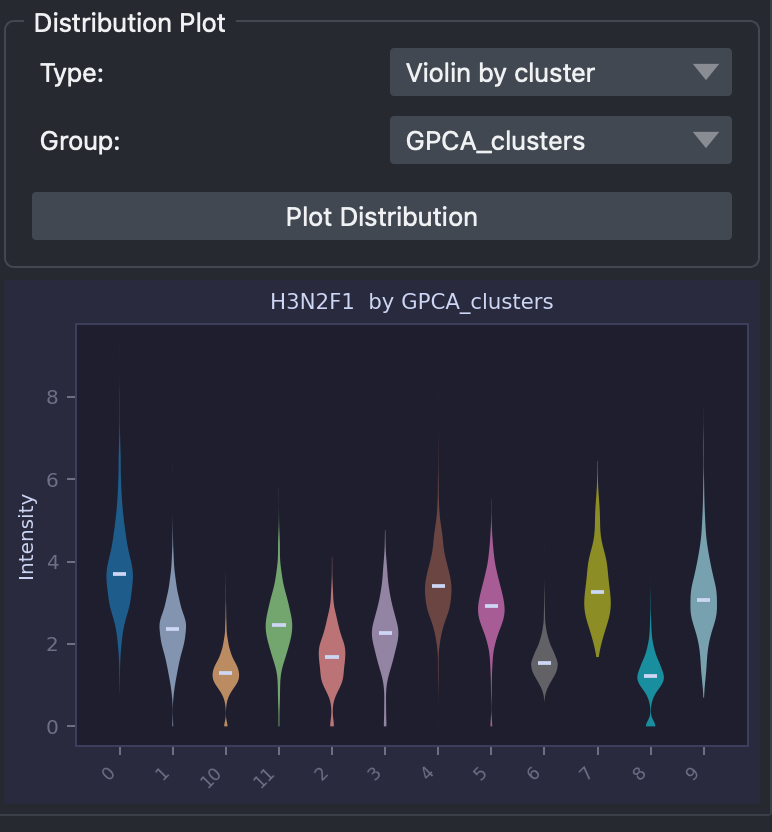
In the section below we can generte some statistical plots for the relative glycan selected. These plots include violin, boxplots or histograms and can be split based on catagorical groups stored in the metadata such as clusters or annotations. For example we will generate a violin plot of glycan ‘H3N2F1’ intensity values for each cluster.
Metadata Plotting
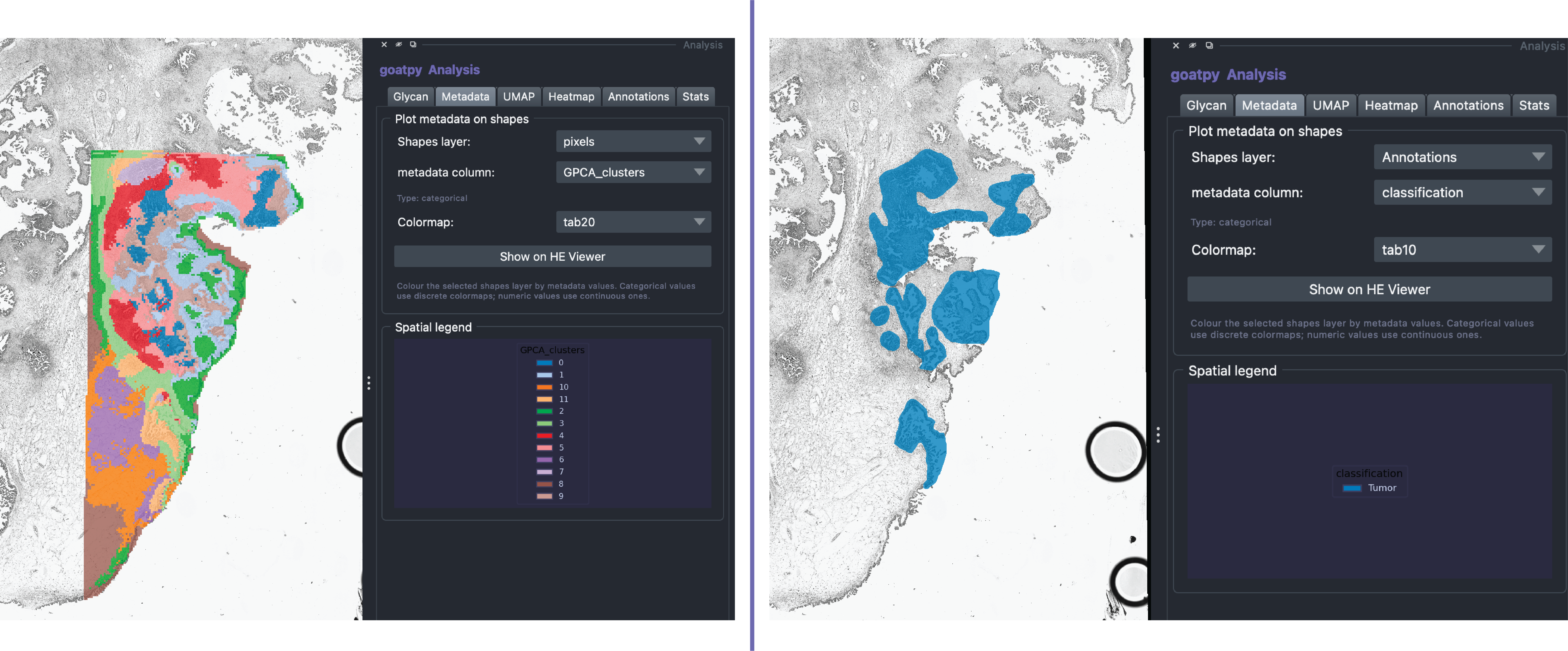
Like the ‘Glycan’ tab, the Metadata tab can be used to visualise various metadata data stored within the goatpy SpatialData object during analysis. For example such as clusters (left) or pathologist annotations (right):
Dimentionality Reduction
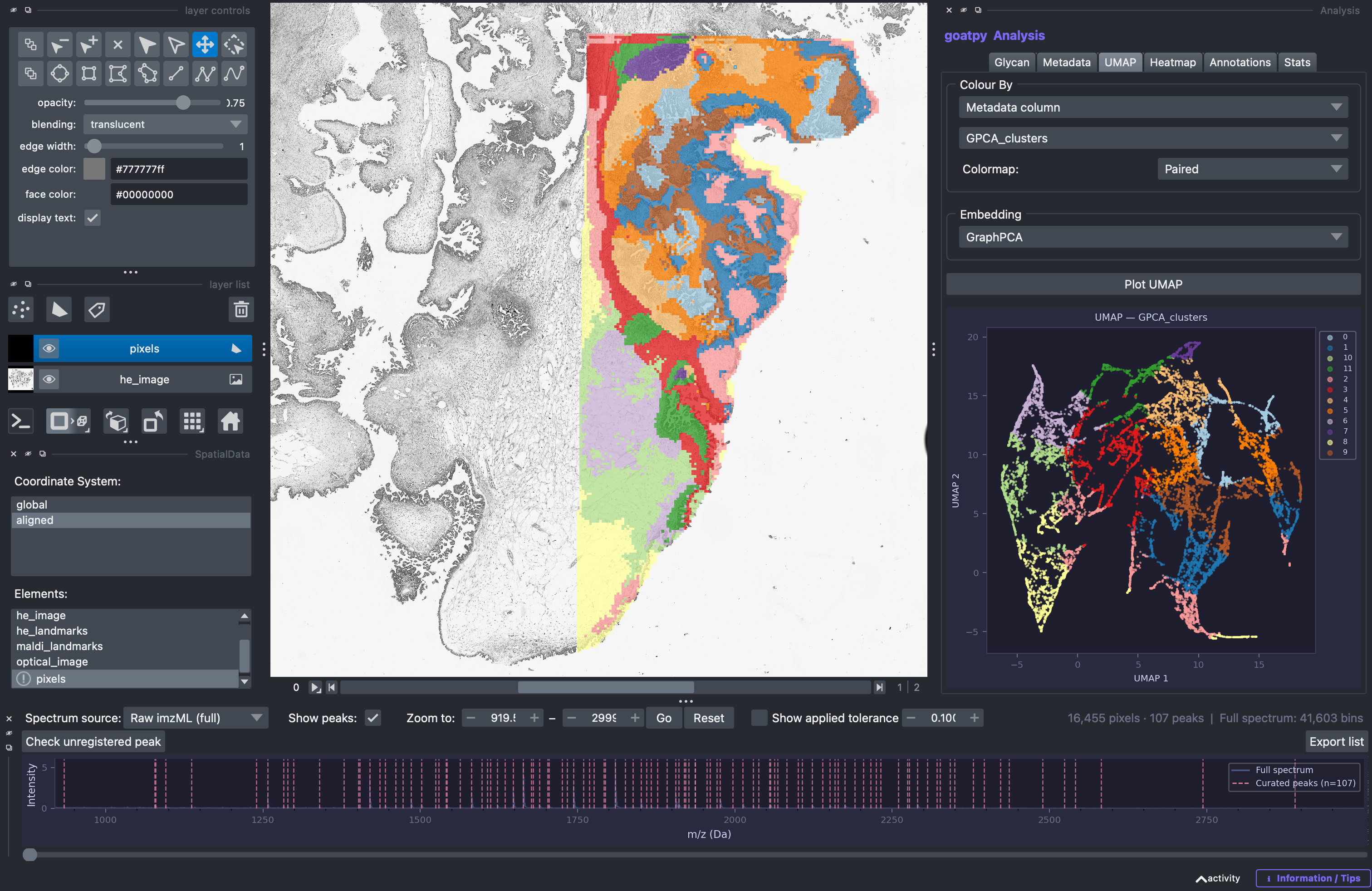
Another key visualisation possible with the goatpy Analysis GUI is visualising dimentionality reduction’s such as UMAP or PCA. This is helpful to compare clusters and distributions of certain glycans across these clusters. Using the UMAP tab we can select which dimentionality reduction we would like to visualise and also what data to overlay onto the plot. For example we can plot our GraphPCA clusters:
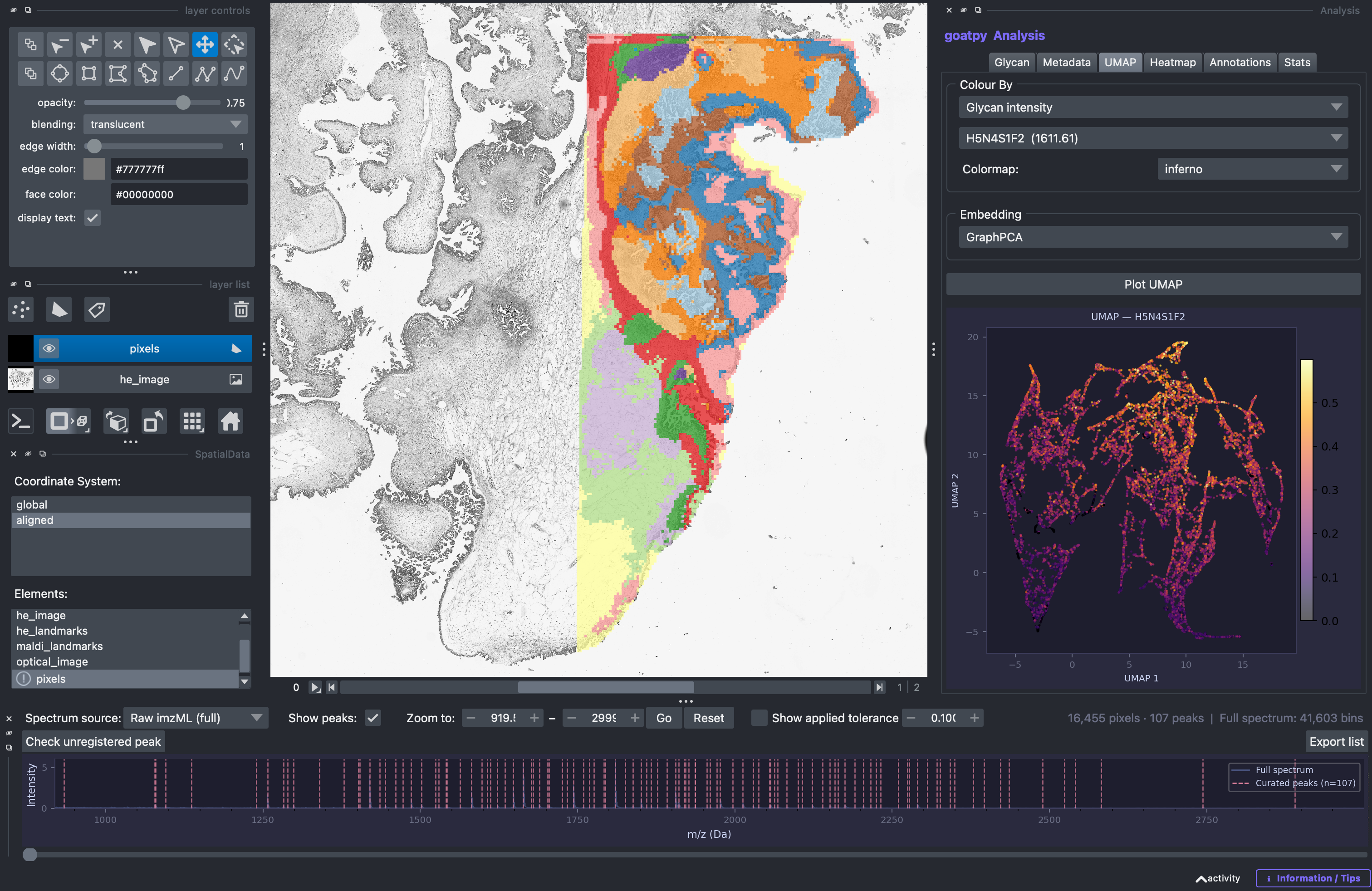
Alternatively, we can also visualise glycan intensity values across this UMAP. By overlaying ‘H5N4S1F2’ (m/z-1611.61) intensity values over the UMAP, we can see that cluster 7 (purple) has the highest abundance of this glycan.
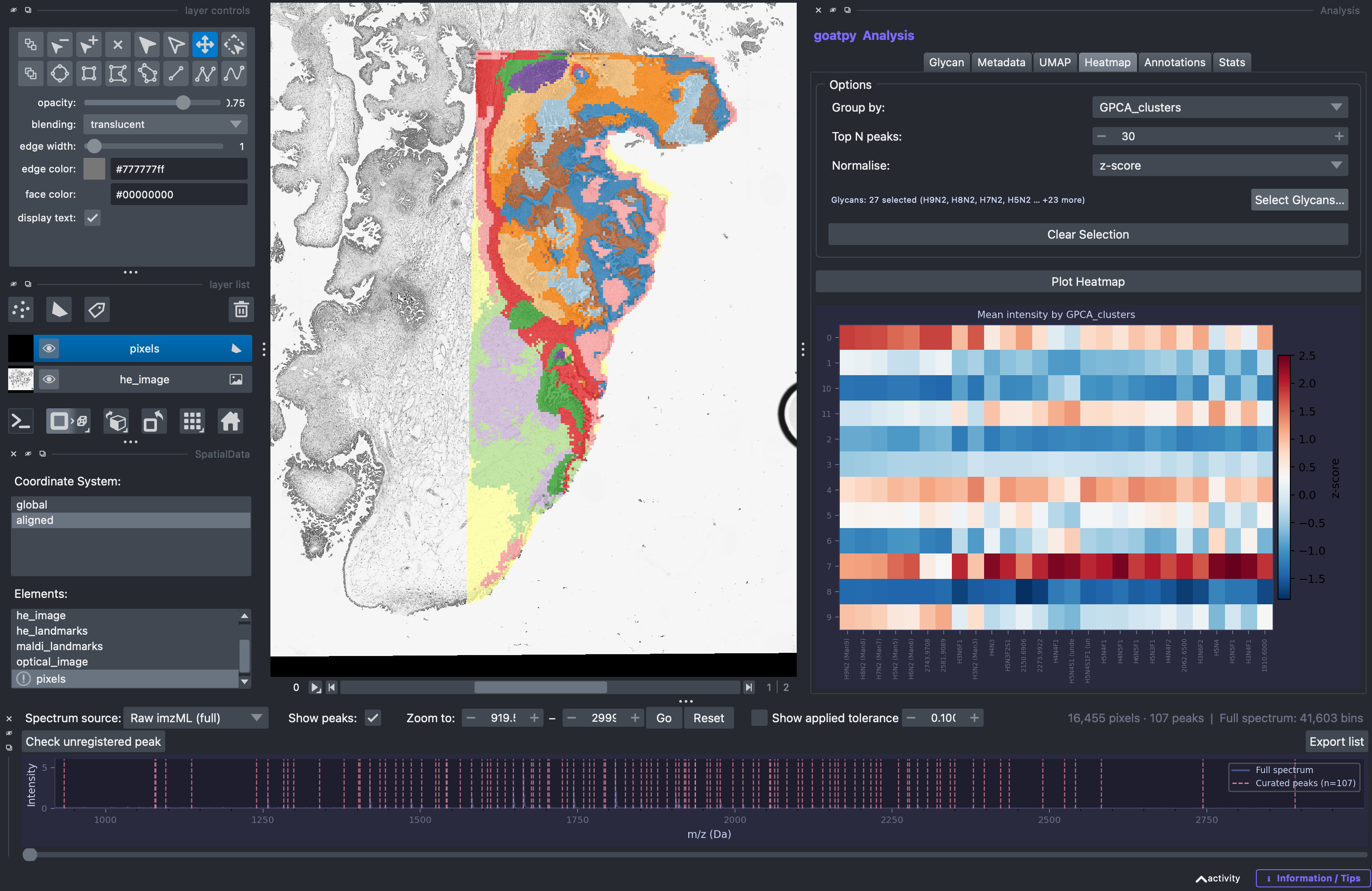
Heatmap Visualisation
Likewise, we can use the Heatmap tab to construct a heatmap to display the mean intensity values of multiple glycans. The heatmap can be grouped by any catagory selected, such as ‘annotations’ or ‘clusters’. In addition we can either use the automatic goatpy function to plot the top n number of glycans (number can be changed) or the user can specify specific glycans they wish to plot. This can be made by hitting the Select Glycans button and by either - selecting specific glycans from
the list, typing out individual glycan or by uploading a .csv file.
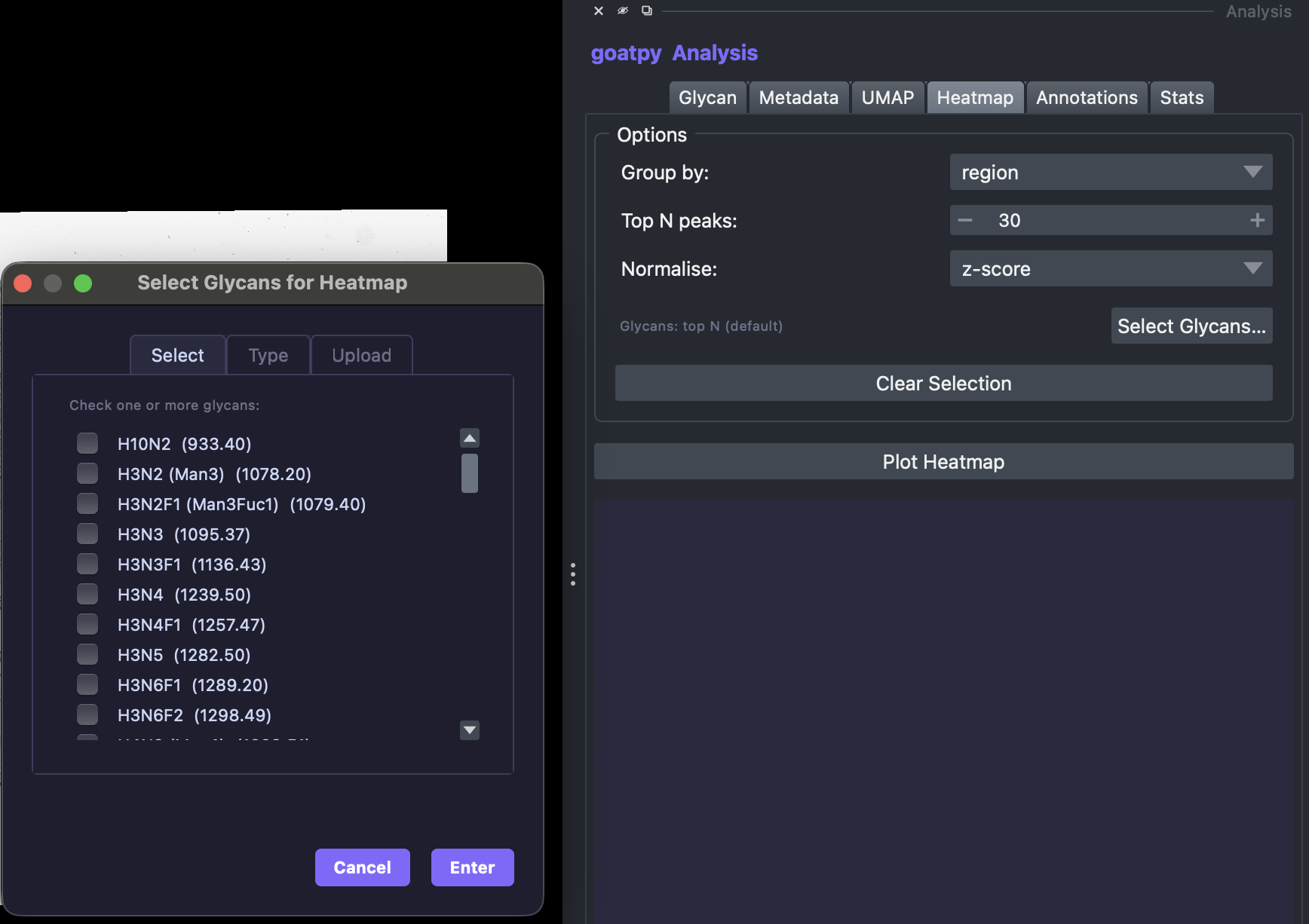
Below we will upload a .csv file with one column (no title) containing a list of glycans which display differential abundance across certain clusters.
Pathologist Annotation Editing
Pathologist annotations based on H&E or IHC images are often critical to any analysis pipeline. goatpy already implements compatability with QuPath exported .geojson files to store these annotations within the data object.
The goatpy GUI lets users visalise, relabel, recolour, edit and add new annotations to the dataset. By selecting the Annotations tab in the Analysis panel users can observe the already assigned annotation:
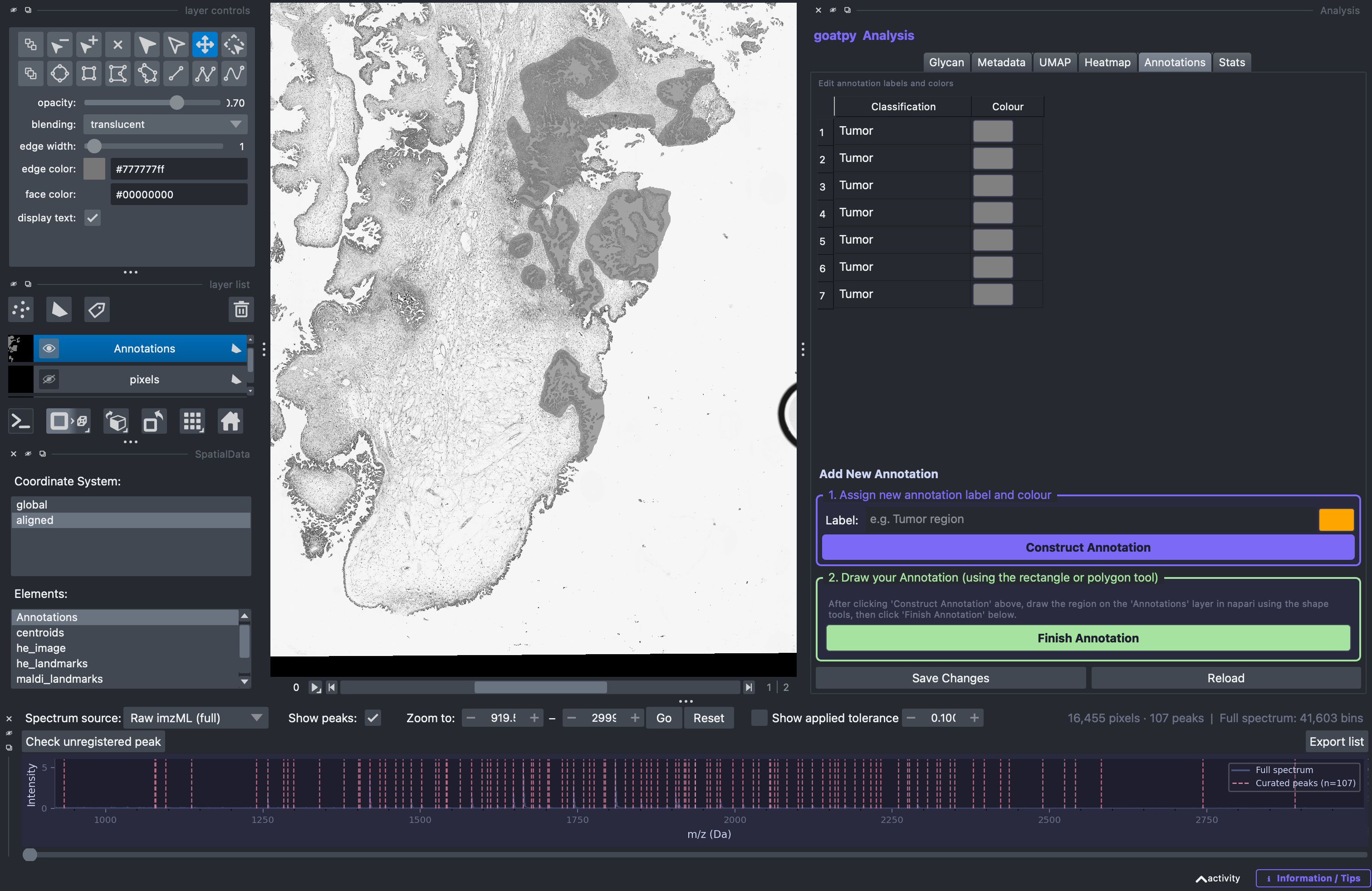
Editing Annotations
By double clicking the name users can edit the label, and by selecting the colour users can assign different colours to different annotations:
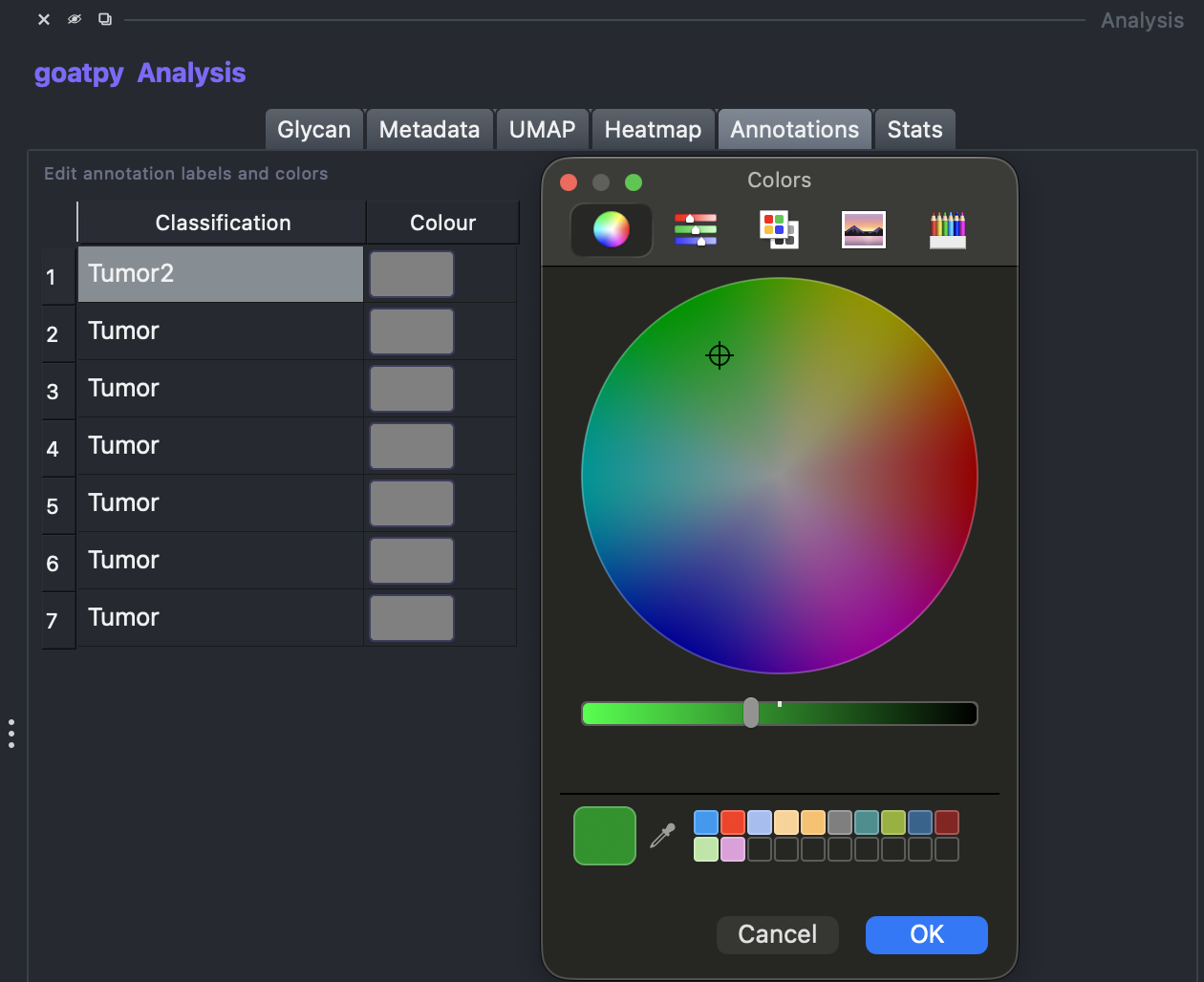
Once finished editing the annotations, hitting the Save Changes button will save these results and display them on the spatial viewer.
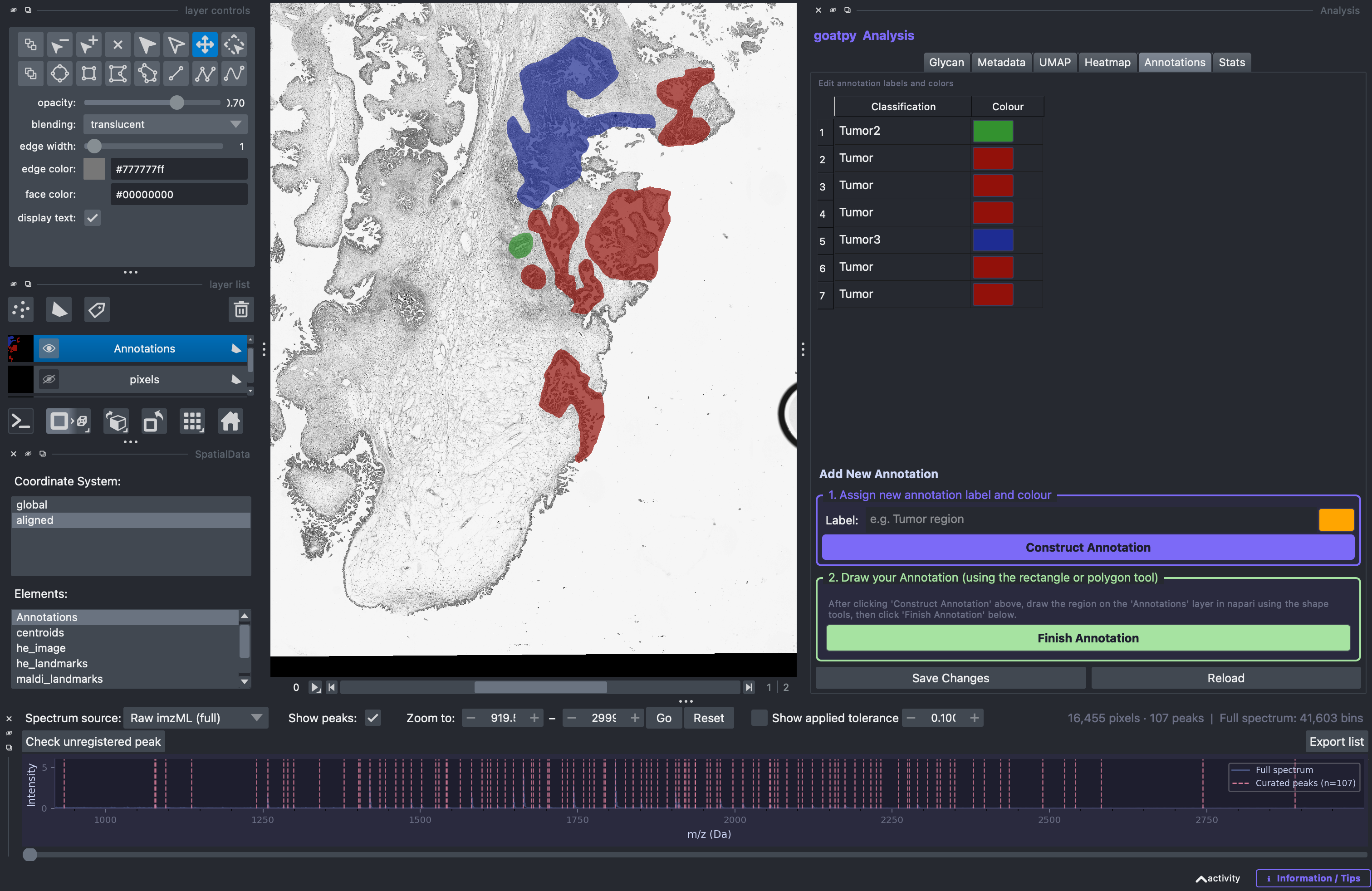
Adding New Annotations
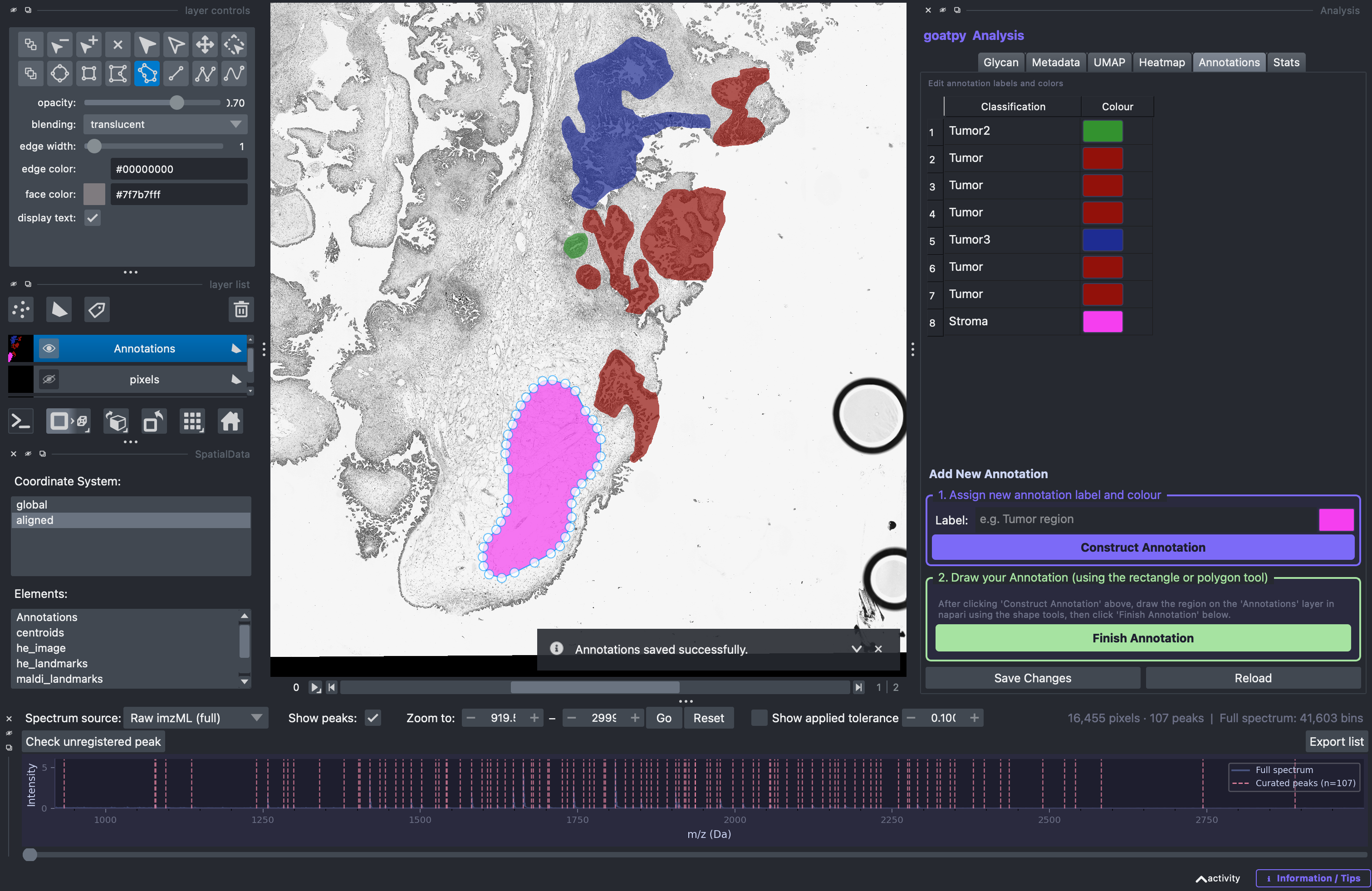
The user can also add new annotations by following the steps in the ‘Add New Annotations’ section (bottom). First, a label must be entered and a colour selected, followed by the Construct Annotation button.
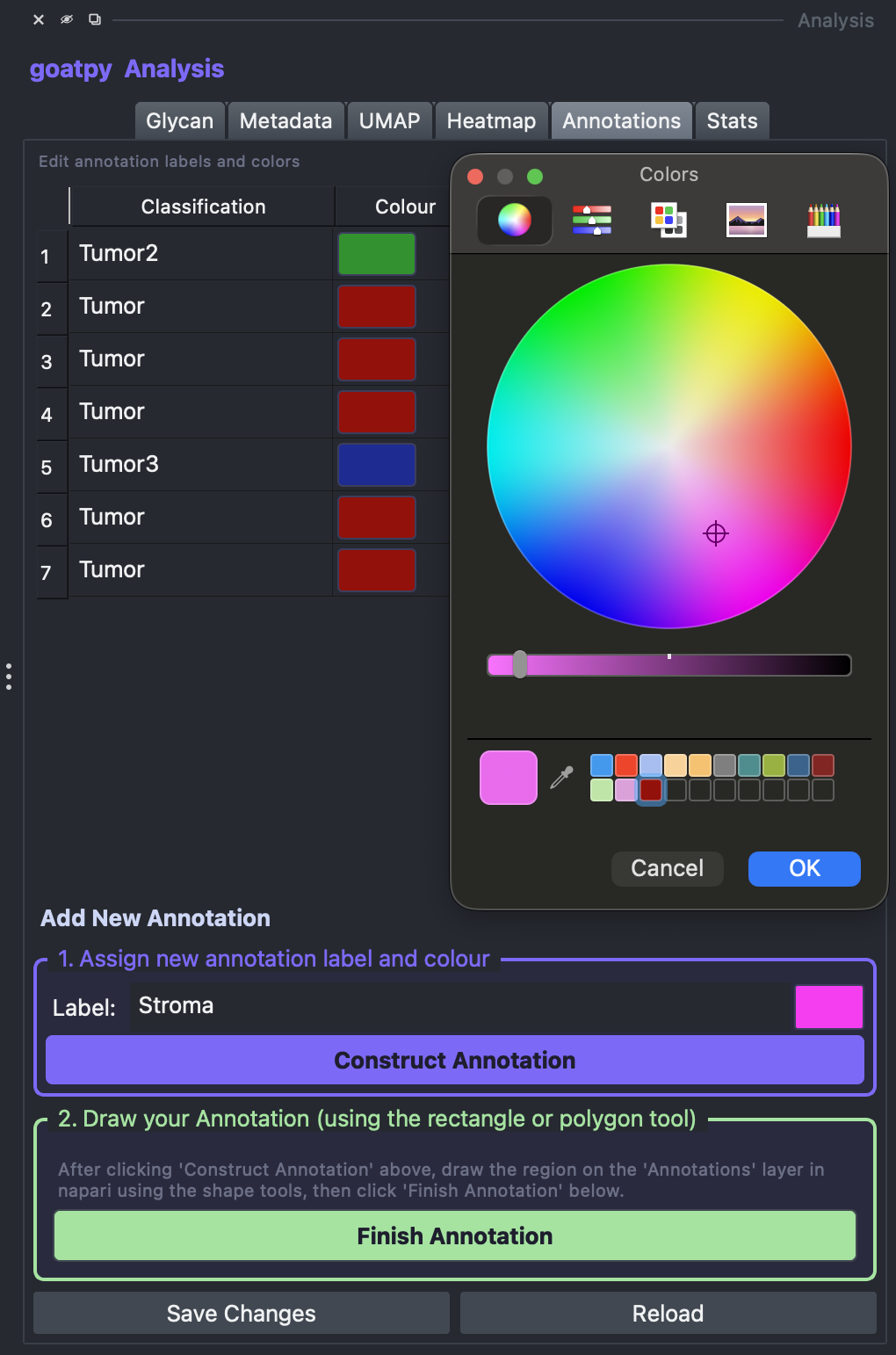
Once done, the new label and colour will appear on the annotation list on the right side. The next step is to head to the Layer Control panel (top left) and select any of the ‘add’ drawing tools (elipse, rectangle, polygon, polygon lasso) to draw the new annotation. Note: ‘add polygon lasso’ tool used below:
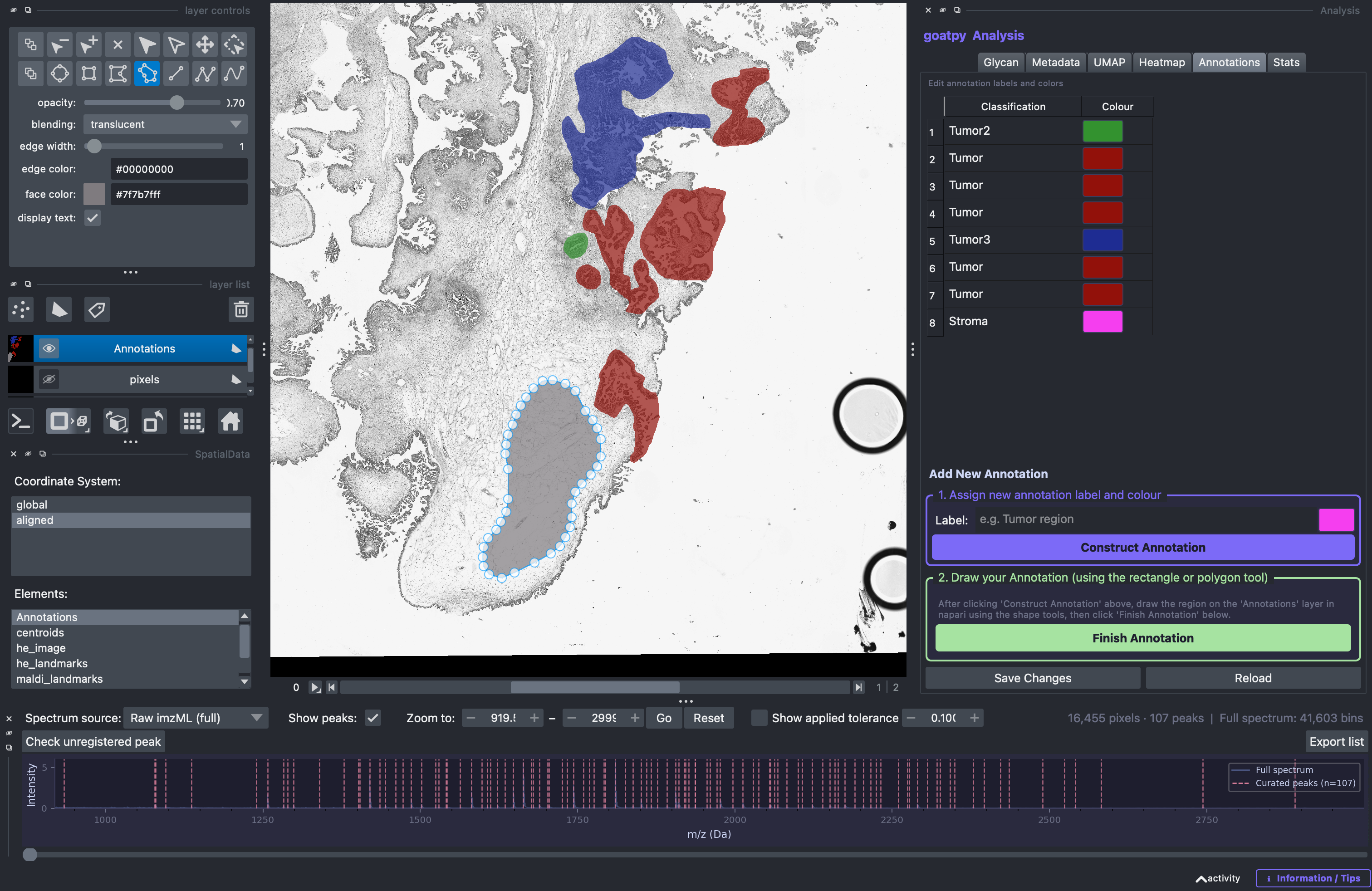
Once satisfied with the polygon outlining the new annotation the Finish Annotation button must be selected. Once done an infomation message like this should appear:
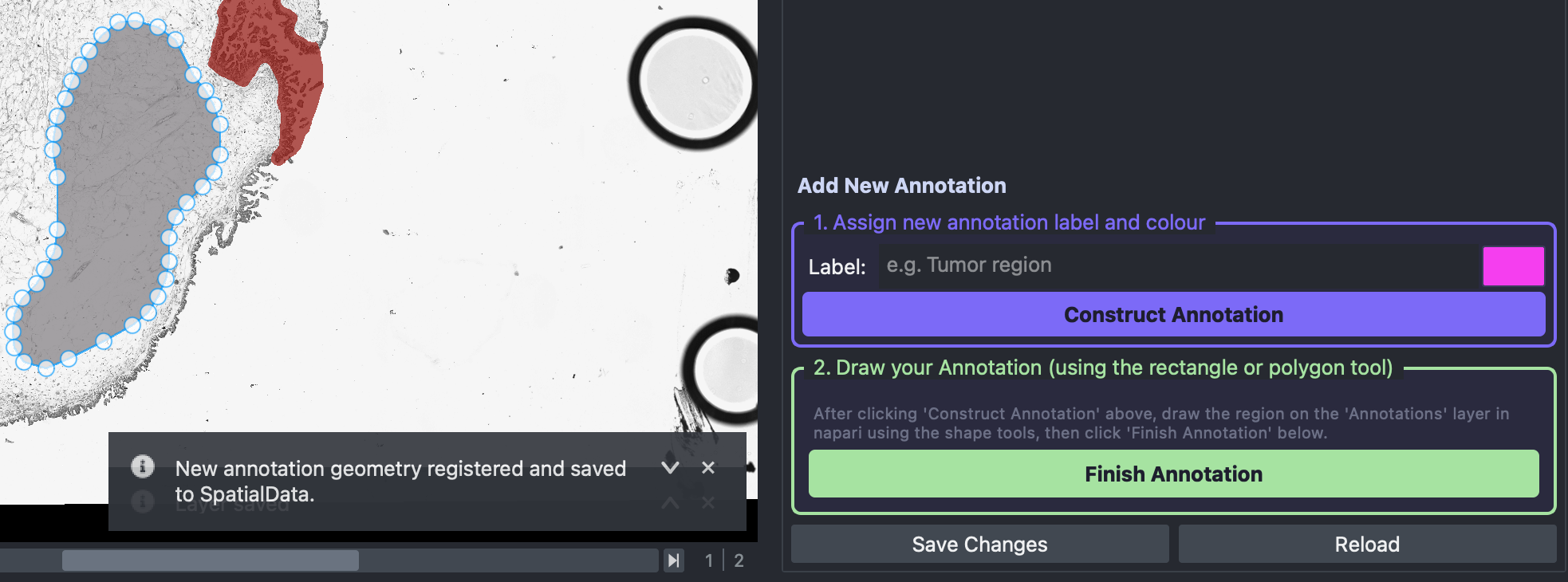
Then the user should select the Save Changes button and the new annotation will be saved and visualised in the spatial viewer.
Saved changes will be updated in the goatpy object meaning futher analysis with these new annotations is possible. We can check this by heading back to the ‘Metadata’ tab and plotting the annotations.
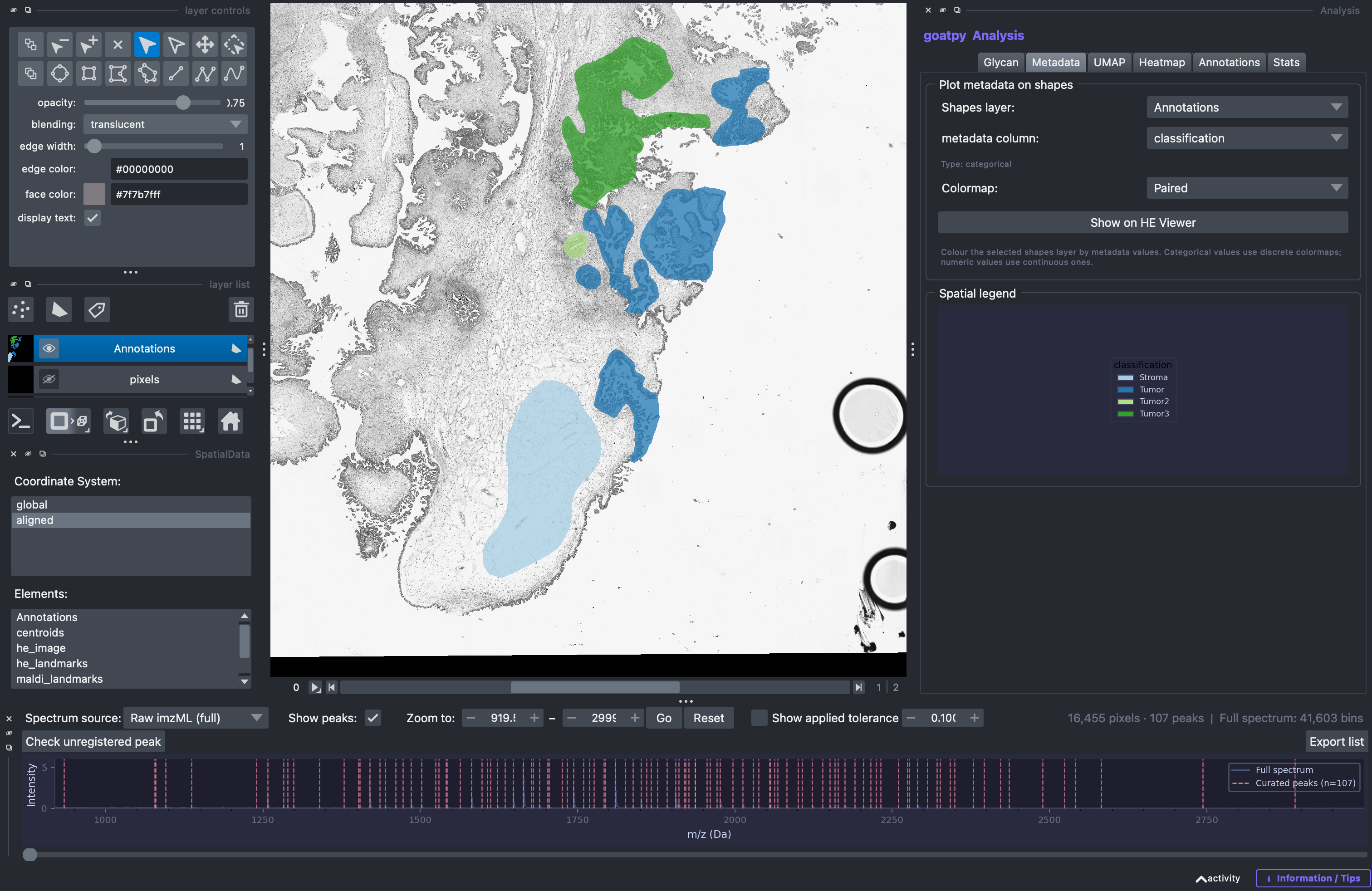
We can see that our new annotation and updated annotation names have been saved successfully.
Naming Glycans (Nomenclature)
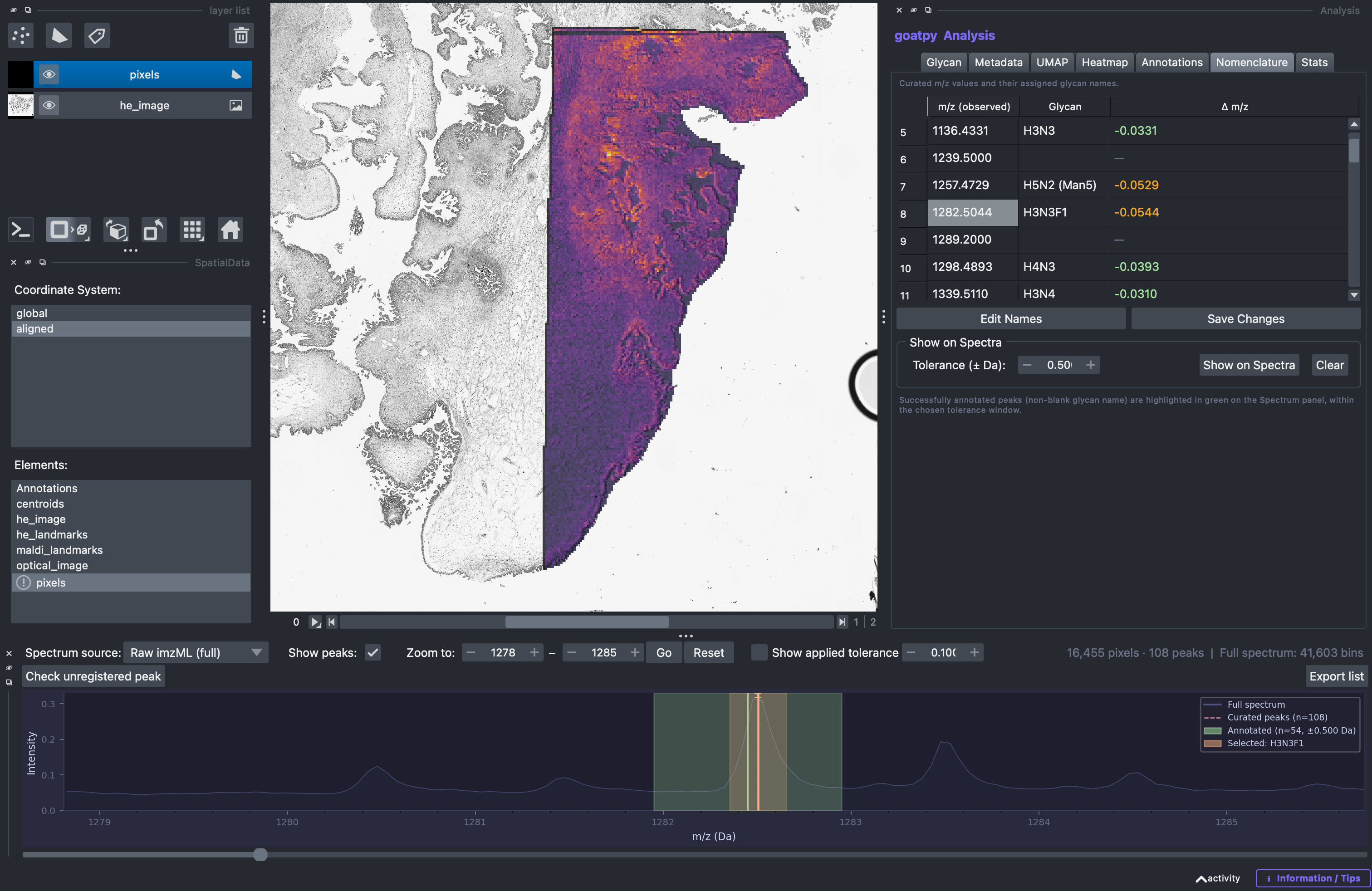
Labeling m/z values with glycan names is import to identify biologically relevant glycans. The Nomenclature tab of provides the user the ability to check, rename and add new glycan labels to each m/z value present within the dataset. In addition, by selecting the m/z value of interest, zooming in on the spectrum, and clicking the Show on Spectra button, we can visualise the m/z range over which the selected peak was assigned its corresponding glycan annotation.
General Statistics
The final tab of the ‘Analysis’ panel is the Stats section which contains some general statistics and plots to highlight the quality and spread of the data.